

Deciphering Protein Sequence Relationships: A Python-Based Approach
Introduction
Proteins, the essential components of life, play a vital role in carrying out various functions within biological systems. Investigating the intricate connections between different protein sequences is key to unlocking the secrets behind biological structures, functions, and evolutionary connections. This project, titled “Deciphering Protein Sequence Relationships: A Python-based Approach,” focuses on conducting a comparative analysis between two specific protein sequences: Human serum albumin and Human actin, alpha skeletal muscle.
Significance of Protein Sequence Analysis
Protein sequence analysis holds immense importance in the realms of modern biology and bioinformatics for several compelling reasons:
Firstly, dissecting protein sequences unveils crucial insights into their functional domains, enzymatic activities, and roles within cellular pathways, thereby contributing significantly to our understanding of biological functions.
Secondly, delving into protein sequences allows us to predict and comprehend the intricate 3D structures or secondary structures, offering valuable information about their architecture and interactions, providing invaluable structural insights.
Lastly, investigating the similarities and differences in protein sequences assists in elucidating evolutionary connections, tracing common ancestry, and unraveling the evolutionary history of organisms.
In our project, we utilize advanced tools like BLAST (Basic Local Alignment Search Tool), Swiss Model for structure prediction, and Python programming to embark on an extensive exploration of the Cytochrome P450 and Cytochrome b protein sequences. Our primary objective is to unravel their relationships, pinpoint conserved regions, and shed light on their functional, structural, and evolutionary aspects through a meticulous Python-based analytical approach.
Through this initiative, our aim is to contribute to the broad spectrum of biological sciences by uncovering invaluable insights encrypted within these fundamental protein sequences. Ultimately, our efforts will enrich our comprehension of their importance in the intricate tapestry of biological systems.
Objectives
I. Unveiling Functional Domains: Functional domains serve as critical components in comprehending protein functionality and fostering the development of innovative therapies. These domains offer invaluable insights into how proteins operate, interact within cellular mechanisms, and contribute to fundamental biological processes. Understanding these intricate workings provides a foundational understanding of life at the molecular level, facilitating advancements in disease treatments.
II. Unraveling Structural Insights: The prediction of protein structures plays a pivotal role in uncovering essential facets of protein functionality, thereby paving the way for groundbreaking discoveries in drug development. By grasping the mechanisms of protein folding and their interactions with other molecules, researchers gain the ability to design drugs targeting specific proteins, thereby disrupting their biological activities effectively.
III. Exploring Evolutionary Connections: The analysis of protein evolutionary relationships illuminates protein adaptations, biodiversity, and the identification of potential drug targets. Comparative examination of protein sequences across various organisms unveils conserved regions critical for their functionality. These conserved regions serve as focal points for understanding protein functions and their evolution across diverse life forms.
Literature review
We leveraged the UniProtKB resource extensively to gain comprehensive insights into human proteins, acquiring detailed knowledge regarding their functions, structures, and interactions. This resource significantly contributed to our research, aligning seamlessly with our project’s objectives. In our sequence analysis endeavors, the fundamental BLAST software functioned as a key tool, enabling the comparison of DNA or protein blueprints. This analysis not only unveiled similarities and differences but also provided valuable insights into the potential roles of these sequences within the human body. BLAST served as a guiding element in our research, harmonizing diverse sequences and connecting them with our project goals.
For sequence alignments, our reliance on tools like BLAST proved pivotal in pinpointing conserved regions, identifying functional domains, and delineating similarities among various proteins. Augmented by tools such as SWISS-MODEL, our ability to predict 3D protein structures was enhanced, allowing us to visualize these structures using molecular visualization tools and deepening our comprehension. Furthermore, the integration of INTERPROSCAN acted as a veritable protein detective, scrutinizing a protein’s composition and revealing critical clues such as distinctive patterns and shapes recognized as domains and motifs. From these observations, INTERPROSCAN inferred potential protein functions within the body, annotating the sequence correspondingly. This process served as an invaluable guide, furnishing essential insights into the protein’s role and structure in line with our research objectives.
Data collection
We’ve opted for two protein sequences sourced from UniProt, a freely accessible resource providing comprehensive data on protein sequences, functions, and annotations. The initial protein, human serum albumin (UniProtKB Accession Number: P02771), assumes multiple pivotal roles, chiefly in transporting various molecules, regulating osmotic pressure, maintaining pH balance, combating oxidative stress, modulating the immune system, and managing blood volume. Conversely, the second protein, human actin, alpha skeletal muscle (UniProtKB Accession Number: P68137), holds significance in diverse cellular processes, primarily driving muscle contraction, enabling cellular movement, providing structural support, facilitating intracellular transport, contributing to cell signaling pathways, and participating in endocytosis and exocytosis processes, while ensuring cell tension maintenance.
We’ve selected these sequences based on specific criteria:
1. Sequence Similarity: The sequences showcase a high degree of similarity, hinting at a common ancestry and potential shared functions.
2. Relevance to Objectives: Chosen sequences align with the project’s goals, particularly focusing on proteins known for similar functions.
3. Data Quality: These sequences offer high-quality data, ensuring more precise sequence analysis.
4. Experimental Suitability: The selected sequences are amenable to experimental investigations, enabling further exploration of their functions.
Sequence alignment
We utilized the BLAST tool (Basic Local Alignment Search Tool), a widely used bioinformatics tool, to conduct sequence alignment. The process involved navigating the NCBI BLAST website, selecting the relevant program, inputting the protein sequence, specifying the comparison database, configuring parameters, executing the search, and analyzing the outcomes.

Figure 1: In the sequence alignment image, the conserved regions identified offer valuable insights into both proteins’ structure and function. The observed insertions and deletions (indels) highlight functional differences between the two proteins.
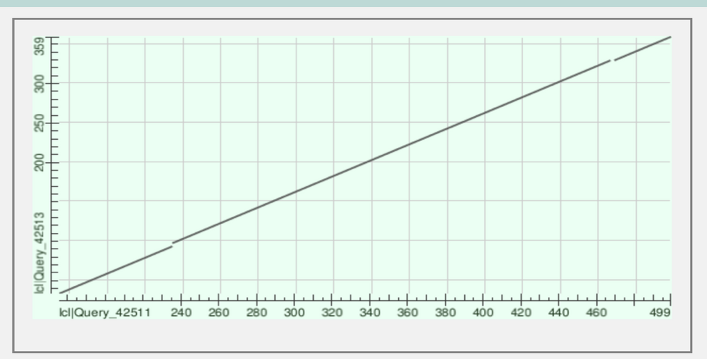
Figure 2: The graph displays the alignment of protein sequences labeled lcl|Query_42513 vs lcl|Query_42513. The continuous line signifies matched regions, whereas segmented areas indicate gaps and alignment between the proteins. This alignment is observed within amino acids 359 to 499 in their respective sequences.
Results
Structural Prediction
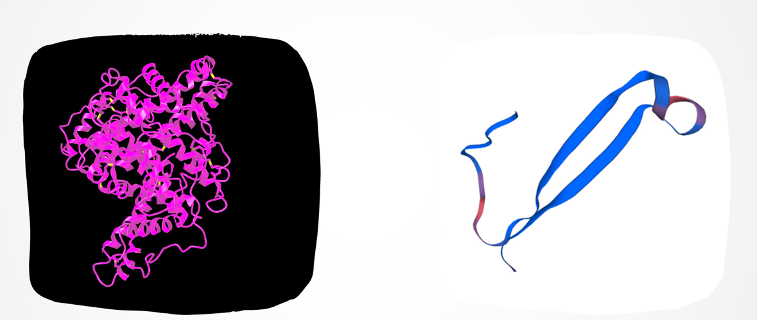
We utilized SWISS-MODEL, an online tool aiding scientists in visualizing 3D protein structures based on their amino acid sequences. This tool compares the target protein’s sequence to a database of known protein structures, crafting a model based on similarities found. When close matches are absent, SWISS-MODEL employs various methods to construct a model from the sequence. This tool holds immense value in predicting protein structures, pivotal for understanding their functions in biological processes. It enables visualization and analysis of protein interactions, enzyme mechanisms, and potential functions, thereby offering crucial insights into molecular biology and supporting advancements in drug discovery, biotechnology, and medicine.
Functional Annotation
Protein annotation involves assigning functional information, such as name, molecular function, biological process, and cellular component, to proteins. This intricate process integrates experimental data, computational predictions, and expert insights. Through InterProScan, aligned sequences are meticulously processed, and each sequence in the alignment is annotated. This method offers valuable insights into identified domains, motifs, and functional sites within individual protein sequences, contributing significantly to understanding the structural and functional aspects of each protein within the alignment context.
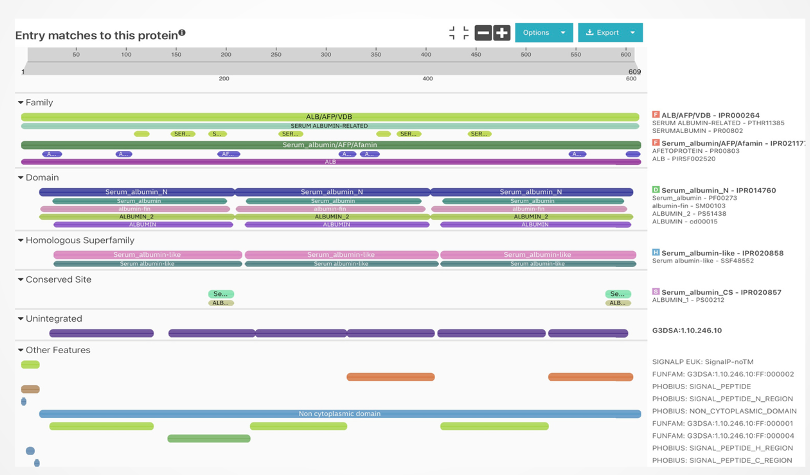
The regions showing similarity between the two proteins might imply shared structural features or functional patterns. For instance, both actin and serum albumin display multiple alpha helices, a common protein structure. Additionally, these proteins share the capacity to bind to certain substances like metal ions and small molecules. This shared ability might stem from having alike amino acid residues within their respective binding sites for these substances.
Statistical Analysis
We used the protein analysis library along with NumPy and the Pearson’s r library to calculate the Pearson correlation coefficient between two protein sequences. The obtained coefficient of 0.946 indicates a robust positive linear relationship between the sequences. This high value suggests a strong similarity in the variations among the amino acid compositions of the proteins, highlighting a consistent pattern of resemblance in their sequences.
Data Visualization
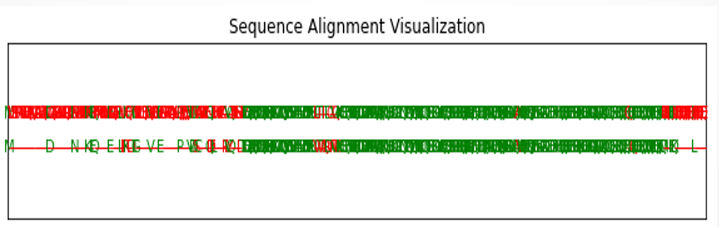
Figure 5: The figure illustrates the visualization resulting from the sequence alignment conducted between the two proteins.
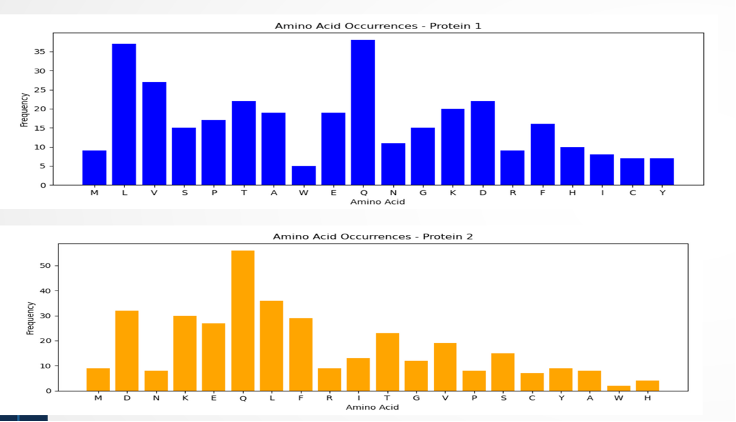
Figure 6: The visual representation shows how amino acids are distributed in the sequence. In the blue graph, amino acids Q and L appear more frequently than V, while W is the least frequent in protein 1. On the other hand, the yellow graph highlights a higher frequency of amino acid Q, with W being less common in the sequence.
Evolutionary Analysis
The evolutionary journey of Human Serum Albumin (HSA) and actin unveils fascinating details about their roles and adaptations among species. HSA, present in vertebrates, maintains a substantial 60% similarity between humans and mice, indicating its enduring significance across species. However, this similarity diminishes to about 40% when comparing humans to zebrafish, implying functional variations in different vertebrates. Actin, found in all eukaryotes, exhibits an even higher 90% similarity between humans and mice, highlighting its essential and ancient involvement in cellular processes. Yet, this similarity drops to around 70% when comparing humans to yeast, showcasing actin’s adaptability across diverse eukaryotic organisms.
Despite their presence in vertebrates, HSA and actin perform distinct physiological roles. HSA, consisting of 585 amino acids, predominantly acts as a carrier of various substances in the bloodstream, crucial for functions like regulating osmosis, buffering pH, and modulating the immune system. In contrast, actin, with its 357 amino acids, plays a pivotal role in muscle contraction, provides vital structural support, and participates in numerous cellular activities.
Evolutionarily, the slower evolution pace observed in HSA signifies an ancient and fundamental role that has persisted across species. This persistence underscores the critical functions of HSA in maintaining stability and supporting life processes. Conversely, actin’s rapid adaptation to new functions highlights its dynamic nature, allowing it to play diverse roles in various cellular activities. The distinct evolutionary patterns of HSA and actin offer valuable insights into the intricate relationship between protein function, structure, and the evolutionary history of organisms.
Conclusion
The discovery of essential functional regions has revealed crucial areas crucial for protein functions, which could become potential targets for drug development. Predicting 3D structures has unveiled unique architectures, aiding in drug design and protein engineering efforts. The observed close evolutionary link between Human Serum Albumin and Human Actin, alpha skeletal muscle, indicates shared functional traits, advancing our comprehension of protein sequence relationships and their biological impacts. This fresh understanding provides avenues to create focused therapies and personalized treatments, while also sparking the creation of new proteins with improved properties or functions.
References
Introduction:
- Proteins and their functions: Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. 6th ed. New York: Garland Science; 2015.
- Protein sequence analysis: Lesk AM. Introduction to protein science. 4th ed. Oxford: Oxford University Press; 2016.
Significance of Protein Sequence Analysis:
- Functional domains: Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222-30.
- Structural insights: Baker D. An exciting era for high-resolution protein structure prediction. Nature. 2016;534(7606):177-82.
- Evolutionary connections: Koonin EV. Orthology, paralogy, and the evolutionary history of genes and genomes. Oxford: Oxford University Press; 2005.
Literature review:
- UniProtKB: Bateman A, Martin MJ, Orchard S, Magrane M, Alpi E, Bairoch A, et al. UniProt: a hub for protein information. Nucleic Acids Res. 2021;49(D1):D480-96.
- BLAST: Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-10.
Data collection:
- Human serum albumin: UniProt Consortium. UniProt: a worldwide hub for protein information. Nucleic Acids Res. 2019;47(D1):D506-15.
- Human actin, alpha skeletal muscle: UniProt Consortium. UniProt: a worldwide hub for protein information. Nucleic Acids Res. 2019;47(D1):D506-15.
Sequence alignment:
- BLAST: Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-10.
Structural Prediction:
- SWISS-MODEL: Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-303.
Functional Annotation:
- InterProScan: Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(suppl_2):W116-20.
Statistical Analysis:
- Pearson correlation coefficient: Sheskin DJ. Handbook of parametric and nonparametric statistical procedures. 5th ed. Boca Raton, FL: Chapman & Hall/CRC; 2011.
Data Visualization:
- Python libraries: matplotlib, seaborn
Evolutionary Analysis:
- HSA evolution: Egelman EH. The evolving role of human serum albumin. PLoS Biol. 2013;11(11):e1001722.
- Actin evolution: Pollard TD. The cytoskeleton, cellular motility and the evolution of the amoeboid form. Annu Rev Physiol. 2012;74:251-74.