

Introduction
Proteins are essential for life, performing various functions vital to our biological systems. Their functions hinge on their intricate three-dimensional structures, shaped by how their amino acid chains fold. Understanding this folding process is crucial to uncovering their roles in health, disease, and interactions within our bodies.
In the quest to decode protein structures, traditional methods like X-ray crystallography and electron microscopy faced hurdles like cost and time constraints. Then came AlphaFold, a game-changing AI model by DeepMind [1]. This innovation uses AI algorithms to predict protein structures incredibly accurately and quickly, solely from amino acid sequences.
This project report explores protein folding’s complex world, highlighting AlphaFold’s pivotal role in deciphering these intricate patterns. It dives into the basics of protein structure determination, AlphaFold’s methodologies, and how its predictions impact drug discovery, genomics, and precision medicine.
Through this report, aim is to showcase how AlphaFold unravels protein folding mysteries, offering unparalleled insights into molecular structures, shaping the future of scientific exploration in this field.
Objective
The project report aims to explore protein folding deeply, with a focus on AlphaFold’s crucial role in decoding complex protein folding patterns. It seeks to achieve two main goals:
1. Understand Protein Folding: Investigate how proteins fold, emphasizing its importance in understanding biological functions.
2. Assess AlphaFold’s Impact: Evaluate AlphaFold’s methods in predicting protein structures, exploring its potential in drug discovery, genomics, and precision medicine, paving the way for future scientific advancements.
Methodology
To investigate protein folding using AlphaFold consists of several pivotal steps. Initially, we collect crucial protein sequences from various sources, essential inputs for AlphaFold’s predictive analysis. Next, employing ColabFold, an implementation of AlphaFold, we predict the intricate structures of these proteins based on their sequences.
Within ColabFold, execute computational processes, allowing AlphaFold to generate accurate predictions. Subsequently, we visualize these predicted protein structures using specialized tools, enabling a clearer understanding of their shapes and arrangements. Following this visualization, we rigorously analyze the predicted structures, assessing their accuracy and potential implications in-depth. Finally, we summarize our findings, discussing the significance of these protein folding insights derived from AlphaFold’s predictions. This systematic approach guides us in unraveling the complex world of protein folding, employing AlphaFold’s predictive capabilities to gain valuable insights into these biological structures.
Basic Concepts
Protein Structure: Proteins, made of amino acid chains, adopt crucial three- dimensional shapes determining their functions and interactions within biological processes. Understanding these structures is vital for comprehending protein functionalities.
AlphaFold: Developed by DeepMind, AlphaFold is an innovative AI-driven system utilizing deep neural networks to predict precise protein structures from amino acid sequences. Its accuracy holds significant implications for structural biology, drug discovery, and bioinformatics.
ChimeraX: A sophisticated tool by UCSF, ChimeraX facilitates the visualization and analysis of molecular structures like proteins, aiding researchers in understanding complex biomolecular systems through user-friendly interfaces and diverse functionalities.
Protein Structure
Protein 1:
- Chaperone protein YscY (UniProtKB ID: >7QIH_1) Length: 274 amino acids
- Sequence: MGHHHHHHGNITLTKRQQEFLLLNGWLQLQCGHAERACILLDALLTLNPEHLAGRRCRIV ALLNNNQGERAEKEAQWLISHDPLQAGNWICLSRAQQLNGDLDKARHAYQHYLELKDHN ESP Protein 2:
- Yop proteins translocation protein X (UniProtKB ID: >7QIH_2) Length: 170 amino acids
- Sequence: GAMGTAQSKRSLWDFASPGYTFHGLHRAQDYRRELDTLQSLLTTSQSSELQAAAALLKCQ QDDDRLLQIILNLLHKV
The two proteins serve distinct functions in cellular processes. Protein 1 acts as a chaperone protein, facilitating the correct folding of other proteins into their functional shapes. Conversely, Protein 2 operates as a protein translocator, aiding in the transportation of proteins across the cell membrane.
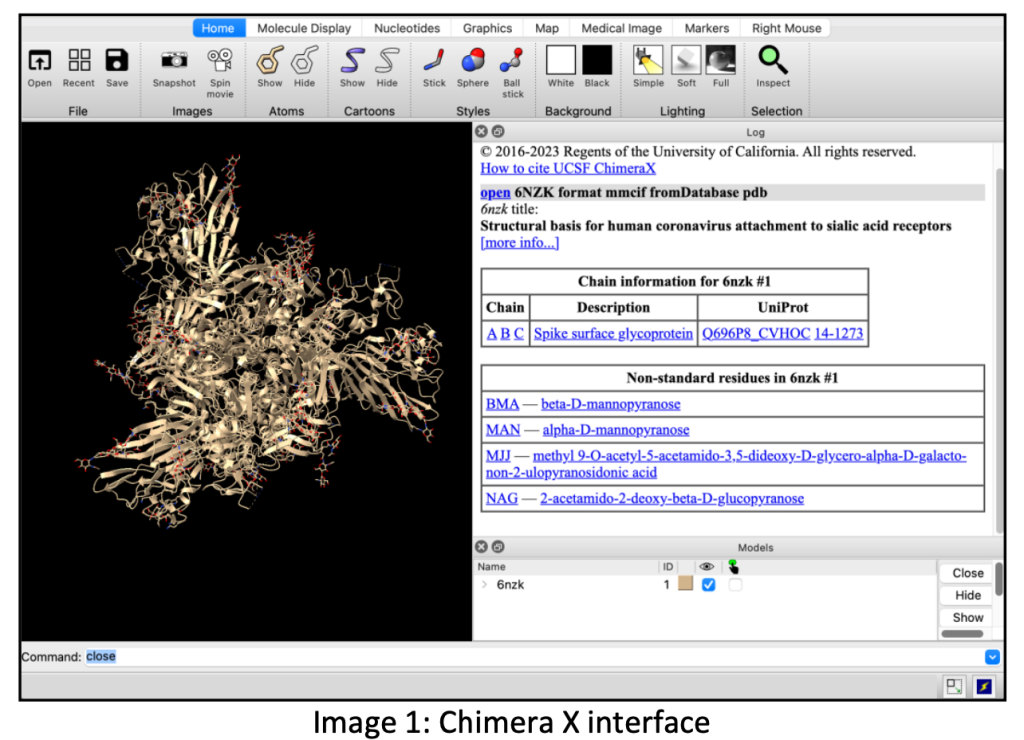
The experimental procedure involves utilizing Chimera X, a visualization and analysis tool, for protein structure prediction. For efficiency, a relatively short protein sequence of approximately 200 amino acids is chosen due to time constraints. Longer sequences are computationally intensive and may take hours to predict, often necessitating the use of supercomputers. When using Chimera X, precision in commands and punctuation is crucial due to its case sensitivity, as errors might alter the results significantly.
The prediction process employs ColabFold, leveraging Google’s GPU support to predict protein structures. While ColabFold expedites predictions, it has inherent limitations, which will be discussed further. Initiating the prediction prompts the installation of ColabFold on the Google Colab virtual machine.
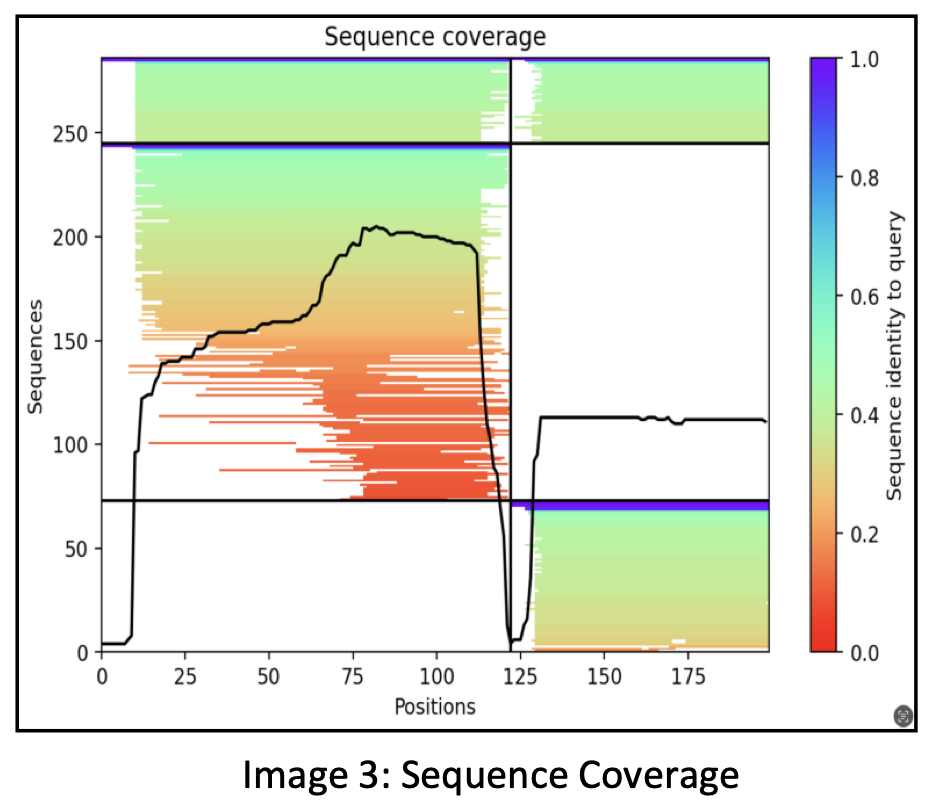
Initially, the process involved sequence alignments where approximately 300 sequences matching the input sequences of the two proteins were identified.
The representation displays a black curve indicating the number of sequences found at each residue position along the horizontal axis of the 200 residues. The division is evident with a black line, delineating the first protein’s 120 residues on the left and the second protein’s residues on the right.
AlphaFold combines these proteins by concatenating them, illustrating the black curve’s coverage for each of these proteins. Approximately 250 sequences cover the C-terminus of the first protein, whereas the coverage for the N-terminus is relatively lower.
In this representation, the colors red, green, and blue signify the sequence identity. Red denotes low sequence identity, green signifies moderate sequence identity, and blue indicates high sequence identity. Sequences displaying higher identity are positioned toward the top of the plot. The white section represents sequences homologous solely to protein 1.
Clarifying the terminologies, the C-terminus refers to the end of the amino acid chain, while the N-terminus marks the beginning of the amino acid chain.

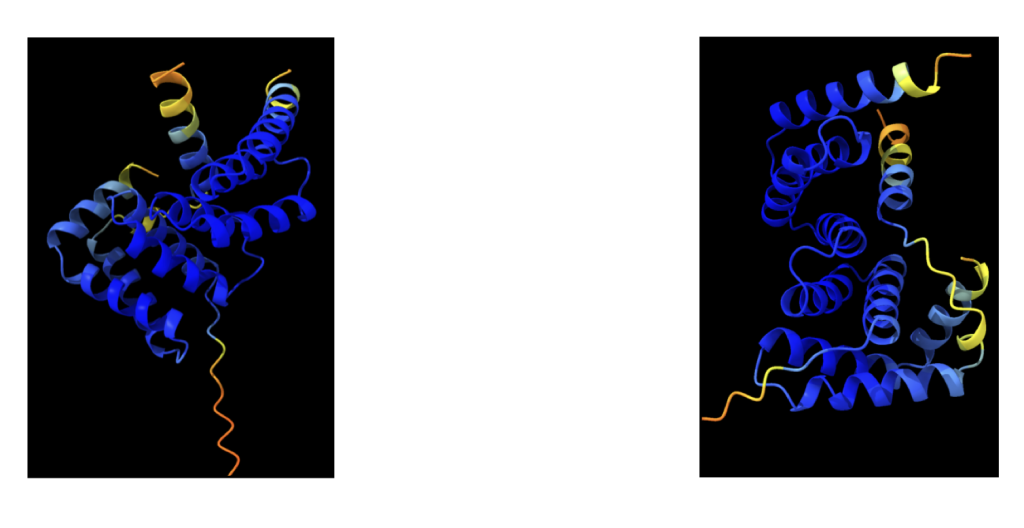
The sequence alignment involves prediction using distinct trained neural networks, resulting in slightly varied predictions. The initial depiction showcases a colored image originating from ColabFold, representing the first predicted complex.
These illustrations portray the intermediate stages of development, revealing slight differences due to the utilization of different neural networks striving to forecast the most accurate structure. Upon running the sequence, all these graphical representations automatically download into the AlphaFold folder.
The model manifests as a three-dimensional shape, employing varied colors to denote prediction certainty. The following criteria help assess the prediction:
- pLDDT (Predicted Local Distance Difference Test): It gauges the model’s fidelity to the distances between neighboring residues in analogous known protein structures. Higher pLDDT scores denote more confident predictions.
- Predicted Aligned Error: This metric measures how closely the predicted model aligns with existing similar protein structures. A lower predicted aligned error signifies a more assured prediction.
- Coloring by Chain: Each protein chain within the model receives a unique color, aiding in distinguishing different domains or regions of the protein. The provided image exhibits a protein model colored based on these criteria. Blue regions represent high pLDDT scores, reflecting high prediction confidence, while white regions indicate low predicted aligned errors, also suggesting high confidence. Differently colored chains facilitate the identification of distinct protein regions or domains.
Significance
AlphaFold catalyzes breakthroughs: revolutionizing protein research, aiding drug development, uncovering disease insights, streamlining studies, contributing to environmental solutions, empowering education, fostering collaboration, advancing genetics, and inspiring innovation. Its predictions drive pioneering strides across biology, medicine, and the environment.
Conclusion
In conclusion, AlphaFold marks a significant leap in structural biology. Its cutting- edge algorithms expedite protein structure determination, surpassing traditional methods in accuracy and speed. This report highlights AlphaFold’s pivotal role in unraveling protein folding complexities, offering insights into functions across various domains, from drug discovery to environmental solutions.
Beyond scientific boundaries, AlphaFold revolutionizes research, fosters collaboration, and inspires innovation in biological sciences. Its contributions promise accelerated discoveries and deeper insights into biological systems.
As AlphaFold continues to redefine protein structure prediction, its impact spans diverse scientific disciplines, paving the way for advancements in biology and medicine. This journey heralds a promising era in unraveling protein folding mysteries, driving scientific progress, and fueling exploration in molecular structures.
Reference
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., … & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589.
- Callaway, E. (2021). Protein-structure prediction explodes: AlphaFold ushers in a new era. Nature, 596(7873), 543-544.
- Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., … & Hassabis, D. (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577(7792), 706-710.
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., & Ferrin, T. E. (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science, 30(1), 70-82.
- Mirdita, M., Ovchinnikov, S., & Steinegger, M. (2022). ColabFold: Making protein structure prediction accessible to all. Nature Methods, 19(2), 153-155.
- AlphaFold website: https://www.theverge.com/2022/7/28/23280743/deepmind- alphafold-protein-database-alphabet
- UniProt website: https://www.uniprot.org/